Our work on mammal evolution is out in Nature!
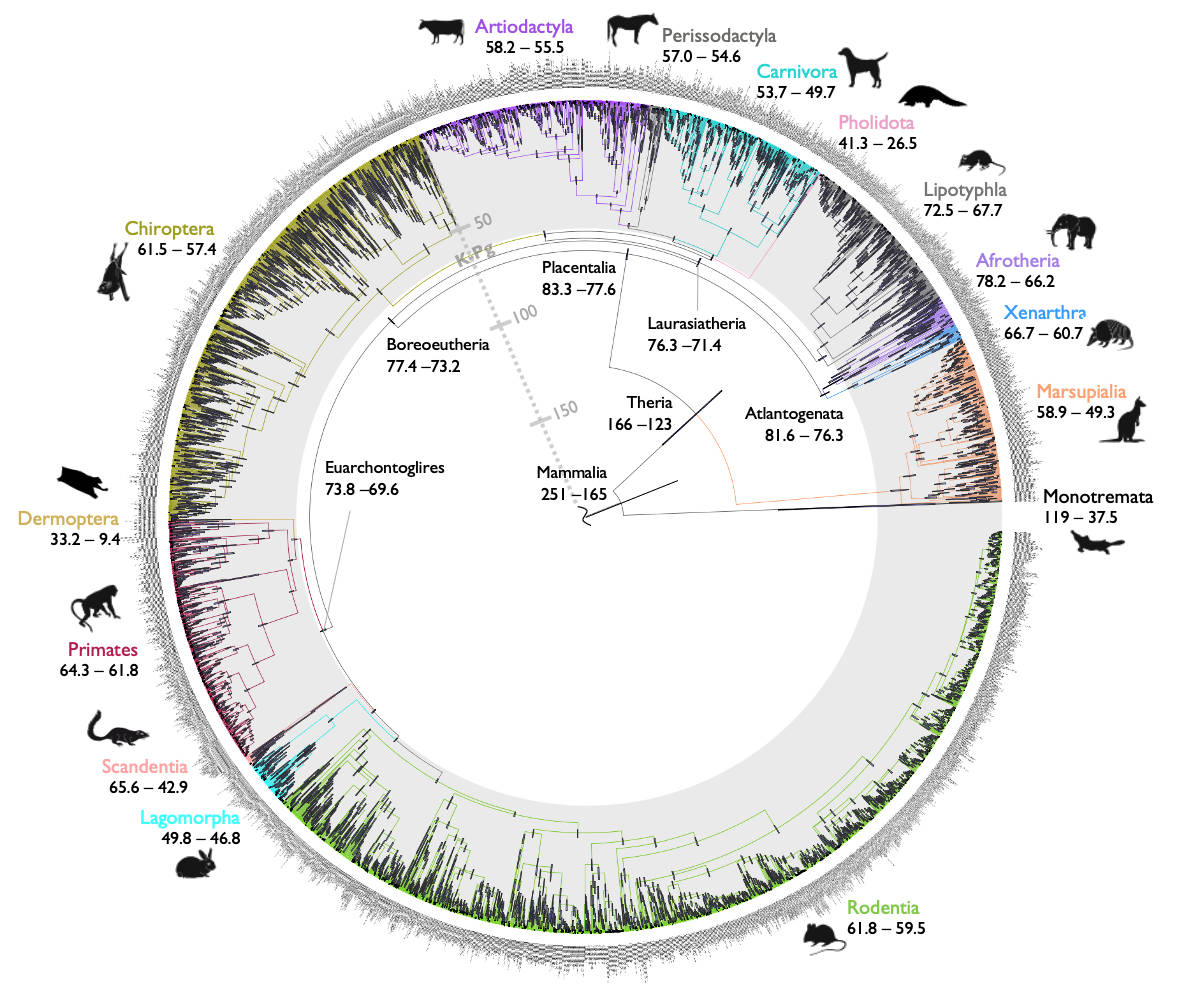
We have estimated a Bayesian timetree of 4,705 mammal species integrating data from 72 genomes. The work was published today in Nature.
Bayesian estimation of evolutionary timelines is extremely expensive because the method relies on stochastic MCMC simulation. The new paper solves the computational hurdles by devising a strategy to separate the analysis into sub-steps. In a first step, MCMC sampling is carried out using the 72 genomes. The posterior distribution of divergence times in this step is then used as a prior to guide MCMC sampling using the full 4,705 species, which are further divided into 13 subtrees.
The use of complete genomes is advantageous because it helps reduce uncertainty in the divergence time estimates. In the case of mammals, this meant we could confidently date the time of origination of crown order groups to the Paleogene, after the K-Pg mass extinction, an issue that has been rather contentious for the best part of two decades.
The methodology we have developed is general and can be used to date other species-level phylogenies integrated with genomic data.
The co-first authors of the study are Sandra Álvarez-Carretero, who carried out the work as part of her PhD at QMUL and who is now at UCL, and Asif Tamuri, also from UCL. The co-corresponding authors are myself, and Phil Donoghue (Bristol University). The other co-authors are Matteo Battini (Bristol University), Fabricia Nascimento (Imperial College), Emily Carlisle (Bristol University), Robert Asher (Cambridge University) and Ziheng Yang (UCL).
The work was funded by the Biotechnology and Biological Sciences Research Council (BBSRC), UK.